

Abstract: Mantle cell lymphoma (MCL) is a rare subtype of non-Hodgkin’s lymphoma
typically marked by an aggressive clinical course and a predilection for relapse. The B-cell receptor (BCR) signaling survival pathway is chronically activated in MCL, contributing to its pathogenesis. Ibrutinib is an inhibitor of Bruton’s tyrosine kinase, a vital component of this pathway. This article details the current clinical experience with ibrutinib in the treatment
of patients with MCL, including completed and published clinical trials and reviews potential adverse events (AEs) and pitfalls associated with ibrutinib therapy. Although most AEs experienced by patients treated with ibrutinib are mild, some can be severe and treatment limiting and may be attributed to off-target effects. Ibrutinib is a very promising agent for patients with MCL with notable response rates. However, when used as a single agent, around one third of patients relapse in the first 2 years of treatment. Recently reported combination therapies have shown significant activity. Emerging data evaluating potential mechanisms of drug resistance and the poor clinical outcomes after treatment failure are also discussed. Further understanding of resistance and its implications not only in relapsed disease but in the frontline setting are needed. Investigation of strategies to overcome resistance remains an area of high unmet clinical need. Evaluation of the impact of shorter treatment duration, effects on minimal residual disease, and incorporation of novel combinations are also warranted.
Keywords: adverse events, ibrutinib, mantle cell lymphoma, resistance
Ther Adv Hematol
2015, Vol. 6(5) 242–252
DOI: 10.1177/ 2040620715592569
© The Author(s), 2015. Reprints and permissions: http://www.sagepub.co.uk/ journalsPermissions.nav
Introduction
Mantle cell lymphoma (MCL) is a rare subtype of non-Hodgkin’s (NHL) B-cell lymphoma defined by cyclin D1 overexpression or t(11;14). MCL often follows an aggressive clinical course with an overall poor prognosis.Young, fit patients are typ- ically treated with chemoimmunotherapy com- monly followed by autologous stem-cell transplant (SCT) [Geisler et al. 2008; Romaguera et al. 2010; Merli et al. 2012; Le Gouill et al. 2014]. However, the average age of a patient diagnosed with MCL is 68 years [Zhou et al. 2008], a group in whom medical comorbidities often play a role during therapy. Therefore, alternative and more tolerable chemoimmunotherapeutic regimens such as bendamustine and rituximab (BR) [Rummel et al. 2013; Flinn et al. 2014] or rituxi- mab, cyclophosphamide, doxorubicin, vincris- tine, and prednisone (RCHOP) with rituximab maintenance [Kluin-Nelemans et al. 2012] are
commonly used for frontline therapy in this pop- ulation. Despite high overall response rates (ORRs) with initial therapy, most patients will eventually relapse and die from their disease. Over the past decade, clinical outcomes appear to have improved, with studies suggesting improved overall survival (OS) among certain cohorts [Herrmann et al. 2009; Chandran et al. 2012]. However, despite the incorporation of aggressive upfront therapeutic strategies and introduction of novel agents, no plateau in survival curves has been appreciated and late relapses even after 5 years of remission can be seen [Romaguera et al. 2010; Geisler et al. 2012; Robak et al. 2015]. There is a clear need for novel and more effective therapeutic options for patients with MCL.
Chronic active signaling via the B-cell receptor (BCR) pathway has been implicated in the pathogenesis of many subtypes of B-cell
malignancies, including MCL. Overexpression of an integral protein in the BCR pathway, Bruton’s tyrosine kinase (BTK), has been observed in MCL cell lines and patient samples [Rinaldi et al. 2006; Cinar et al. 2013]. Activation of BTK and its downstream targets plays a vital role in normal and malignant B cells, including modulation of nuclear transcription, as well as regulation of B-cell proliferation, differentiation, survival and migration [Herrera and Jacobsen, 2014]. In MCL, phosphoproteomic analysis of cell lines indicate a prosurvival role of BCR sign- aling in this malignancy [Pighi et al. 2011]. The importance of BCR signaling pathway proteins in B-cell malignancy pathogenesis has driven interest in using inhibitors of these proteins as therapeutic options.
Ibrutinib is a potent, orally bioavailable inhibitor of BTK that binds irreversibly to a cysteine residue (C481) in the BTK active site. In preclinical studies in MCL cell lines, ibrutinib demonstrated inhibi- tion of downstream BCR signaling and cell apopto- sis[Cinaretal.2013].Inpreclinicalanimalstudies, eight dogs with spontaneous canine B-cell lym- phoma were safely treated with ibrutinib. Single administration of 2.5–20mg/kg per day was suffi- cient to fully occupy BTK in peripheral blood and tumortissuefor24h.Threedogsobtainedapartial response (PR) and three dogs maintained stable disease (SD) [Honigberg et al. 2010].With preclini- cal safety data completed, ibrutinib moved into clinical trials in humans.The purpose of this review is to discuss the clinical experience using ibrutinib inpatientswithMCLandtoidentifypotentialpit- falls, therapeutic challenges, and opportunities in the era of targeted agents. Given its impressive clin- ical activity, ibrutinib has now been approved by the US Food and Drug Administration (FDA) for relapsed/refractory MCL, chronic lymphocytic leukemia (CLL) and Waldenstrom’s macroglobu- linemia (WM) and frontline in patients with CLL with del(17p13.1) karyotype and WM.
Clinical experience with ibrutinib in the treatment of patients with MCL
Ibrutinib single-agent therapy in patients with relapsed and refractory MCL
Even in early phase clinical trials, it was clear that ibrutinib brought promising efficacy to patients with relapsed/refractory (R/R) MCL. An initial phase I multicenter study treated 56 patients with R/R B-cell NHL with single-agent ibrutinib via
multiple dosing schemes. Among the nine patients with MCL on this trial, seven achieved response (three CRs). Most adverse events (AEs) were grade 1–2 with the most common AEs of diar- rhea, nausea, fatigue, and myalgia. Grade 3–4 AEs were rare and most commonly included cytopenias (<15%). The dose of 560mg by mouth daily was well tolerated and led to full- target occupancy in a range of individual body weights and therefore was chosen for phase II study [Advani et al. 2013].
In the pivotal phase II trial that led to FDA approval of ibrutinib in R/R MCL, 111 patients with MCL received single-agent ibrutinib (560 mg daily).The median age of the patients in the study was 68 years (range 40–84) and therefore repre- sentative of the typical MCL patient population. The ORR was 68% with a CR rate of 21%. Again, the most common grade 1–2 AEs were diarrhea, nausea, and fatigue. Grade 3–4 cytopenias were less than 20% (most commonly neutropenia). After initial follow-up of 15.3 months, the esti- mated median progression-free survival (PFS) was 13.9 months [95% confidence interval (CI) 7.0–not reached], and the estimated OS was not reached [Wang et al. 2013]. However, with an extended median follow up of 26.7 months, the estimated median PFS was 13 months with approximately one third of patients failing to respond to therapy at 2 years.The median OS was 22.5 months [Wang et al. 2014b].
Prior to ibrutinib’s FDA approval in MCL, 149 patients with R/R MCL were enrolled on an open-label early-access treatment protocol using single-agent ibrutinib.The median age of patients in this study was also 68 years (range 39–90). Of these patients, 58.5% had refractory disease and 66.7% had received at least three prior lines of therapy. Grade 3–4 AEs were observed in 39.6% of the patients, the most common of which was cytopenia (<10%). AEs of particular interest were one patient with a major hemorrhage and one patient with intracranial bleeding. Long-term follow up has yet to be reported; however, two thirds of the patients in the trial were still taking ibrutinib at study completion (US FDA approval) [Martin et al. 2014a].
Ibrutinib combination therapy in patients with
MCL
In order to improve upon the promising efficacy of ibrutinib as a single agent in the treatment of
patients with MCL, many investigators sought to combine this agent with other therapies (Table 1). Rituximab is a monoclonal antibody that targets CD20, which is used widely in CD-20-positive B-cell malignancies. Rituximab has been studied in combination with ibrutinib in patients with several B-cell malignancies, including MCL. In preclinical studies, concerns arose that off-target effects of ibrutinib may limit the effectiveness of rituximab when used in combination. Specifically, preclinical studies in CLL indicate that ibrutinib may decrease the CLL cell surface expression of CD20 [Bojarczuk et al. 2014] and may decrease or ablate the antibody-dependent cell-mediated cytotoxicity of monoclonal antibodies [Kohrt et al. 2014] via off-target inhibition of interleukin- 2-inducible kinase (ITK) in natural killer (NK) cells [Dubovsky et al. 2013]. Despite these con- cerns, clinical trials with the combination moved forward. In a phase II study, 50 patients with R/R MCL were treated with rituximab (375mg/m2 intravenously weekly×4, then monthly for up to 2 years) and ibrutinib (560 mg orally daily). With a median follow up of 6.5 months (range 1–10), the ORR was 87% with 38% CR rate. All patients with SD or progressive disease had a proliferation index (Ki67) of at least 50%. Of the 33 patients with Ki67 less than 50%, the ORR was 100% with a 48% CR rate. These data suggest that this regimen may be more effective in patients without the blastoid variant, which often is associated with high Ki67. However, the clinical and biologic rel- evance of this finding should be interpreted with caution given that a Ki67 cutoff of 50% is high by MCL standards and likely the result of trying to stratify outcomes using a relatively small dataset. Grade 3 AEs were neutropenia (1) and thrombo- cytopenia (1). Notably six patients developed atrial fibrillation (afib) [Wang et al. 2014a]. A similar ongoing phase II trial pairs ibrutinib with ublituximab, a novel glycoengineered anti- CD20 monoclonal antibody in patients with R/R MCL and CLL [ClinicalTrials.gov identifier: NCT02013128]. Early analysis suggests this combination is active and well tolerated in patients with MCL [Sharman et al. 2014].
Chemoimmunotherapy regimens using cytotoxic chemotherapy plus rituximab and ibrutinib is also an area of active study. Bendamustine is an alkylating agent with efficacy in MCL that is com- monly used in combination with rituximab, dem- onstrating ORR greater than 90% [Flinn et al. 2014]. A phase I/Ib study evaluated the safety of bendamustine (90mg/m2 on days 1 and 2 for six
cycles) and rituximab (375 mg/m2 on day 1 for six cycles) with ibrutinib (280 or 560 mg daily indefi- nitely) in 48 patients with NHL. Of these patients, 17 had MCL (previously untreated = 5). No dose- limiting toxicities (DLTs) were observed. The most common grade 3/4 AEs were cytopenias and rash. Among the patients with MCL the ORR was 94% with a 76% CR rate [Maddocks et al. 2015]. An ongoing randomized international study, which recently closed to accrual investigates this combination (followed by 2 years of maintenance rituximab) in patients over the age of 65 with newly diagnosed MCL (SHINE study) [ClinicalTrials.gov identifier: NCT01776840]. Similarly, RCHOP chemotherapy has long been used to treat MCL with significant efficacy [Flinn et al. 2014]. Another phase Ib study attempted to safely combine RCHOP with ibrutinib in 33 patients with treatment-naïve NHL, including five patients with MCL. Of all patients, the most common grade 3 AEs were cytopenias.The group as a whole demonstrated an ORR of 94% with a 72% CR rate. No specific response data for the patients with MCL were included [Younes et al. 2014]. Without a randomized phase III trial for patients with previously treated indolent MCL, it is unclear whether the combination of ibrutinib with either BR or RCHOP is superior. In an effort to define the contribution of ibrutinib to aggres- sive induction strategies, the European MCL Network is sponsoring the Triangle Study which randomizes patients with MCL to one of three arms: RCHOP alternating with rituximab, dexa- methasone, cytarabine, and cisplatin (RDHAP) followed by autologous SCT; RCHOP/RDHAP with ibrutinib followed by autologous SCT with ibrutinib maintenance; and RCHOP/RDHAP with ibrutinib followed by ibrutinib maintenance. Another trial that uses ibrutinib as maintenance therapy for patients with MCL following various induction therapies including BR and RCHOP- based regimens is open for accrual [ClinicalTrials. gov identifier: NCT02242097]. These studies attempt to address how to best use ibrutinib with chemoimmunotherapy regimens.
Lenalidomide is an oral immunomodulatory agent with single-agent activity in R/R NHL, including MCL [Habermann et al. 2009; Goy et al. 2013; Zinzani et al. 2013]. A phase I study for patients with R/R NHL combining lenalidomide with ibru- tinib has been reported. At the date of the last report, 13 patients had been treated (two patients with MCL). At the first dose level (DL1) of lena- lidomide 15 mg and ibrutinib 420 mg, two DLTs 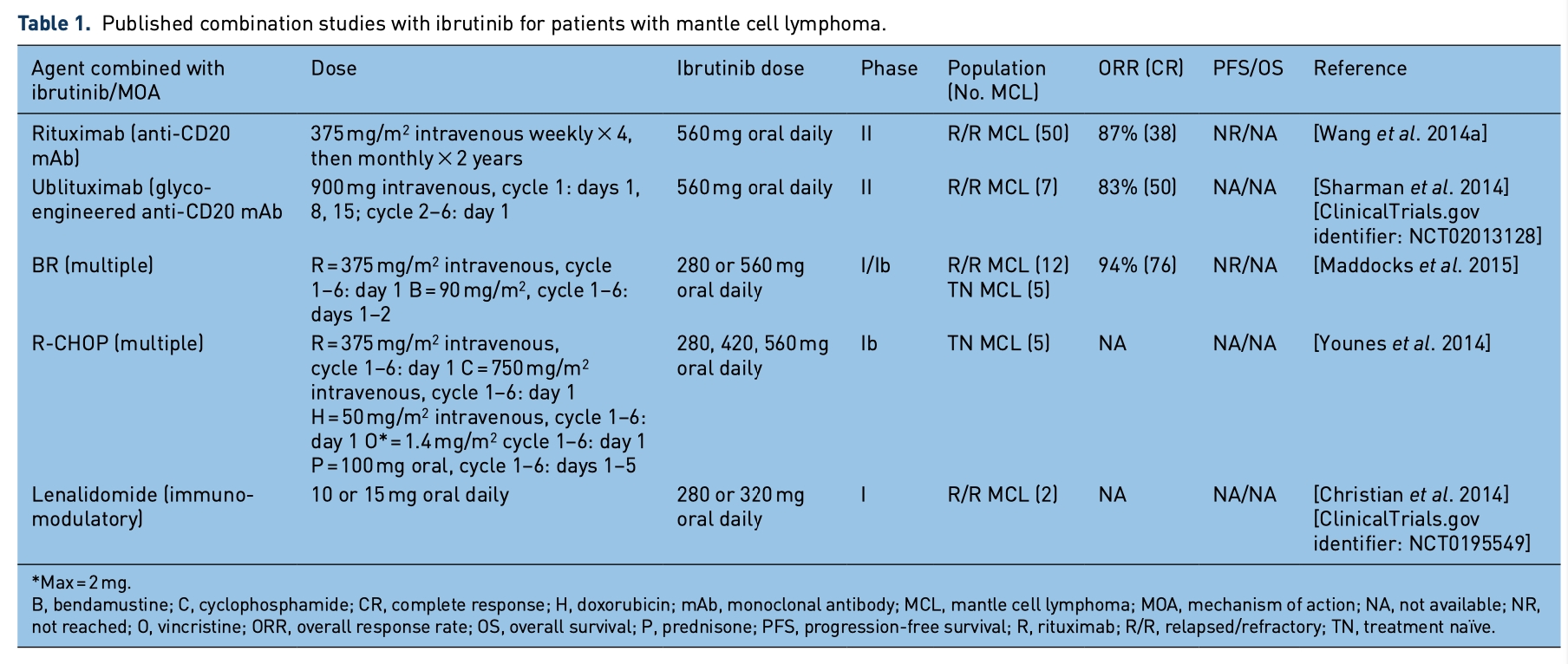
including ischemic stroke and grade 3 rash were observed. Six patients were subsequently treated at a reduced dose level (DL1) of lenalidomide 10mg and ibrutinib 280mg and no DLTs were observed. Grade 3/4 toxicities occurred in 4 of 13 patients (hypokalemia, cytopenias, pneumonia). One of the patients with MCL achieved a PR. An intermediate dose-level trial is currently enrolling [ClinicalTrials.
AEs related to off-target effects of ibrutinib
In addition to inhibition of BTK, ibrutinib can also inhibit other kinases with similar cysteine residues at their active sites, such as ITK, epidermal growth factor receptor (EGFR), tyrosine protein kinase (TEC), B-lymphocyte kinase (BLK), epithelial/ endothelial tyrosine kinase (BMX), human epider- mal growth factor 2 receptor (HER2), and human epidermal growth factor 4 (HER4), among others [Herrera and Jacobsen, 2014; Woyach et al. 2014], which may lead to off-target side effects. ITK nor- mally drives proximal T-cell receptor signaling and positively regulates NK cell’s Fc Receptor-initiated cytotoxicity via recognition of antibody-coated targets [Khurana et al. 2007]. Ibrutinib has been shown to irreversibly inhibit ITK [Dubovsky et al. 2013], which in preclinical models, has been shown to limit the therapeutic activity of monoclonal antibody therapies through ablation of antibody- dependent NK-cell-mediated cytotoxicity [Kohrt et al. 2014]. However, given the impressive clinical activity seen with ibrutinib in combination with rituximab [Wang et al. 2014a] this finding may not be clinically significant. Furthermore, this effect may not be as relevant in the novel Fc-engineered typeIIantibodiessuchasobinutuzumab[DaRoit et al. 2015]. In addition to potentially interfering
gov identifier: NCT0195549] [Christian 2014].
et al.
Other combinations with ibrutinib in patients
with MCL
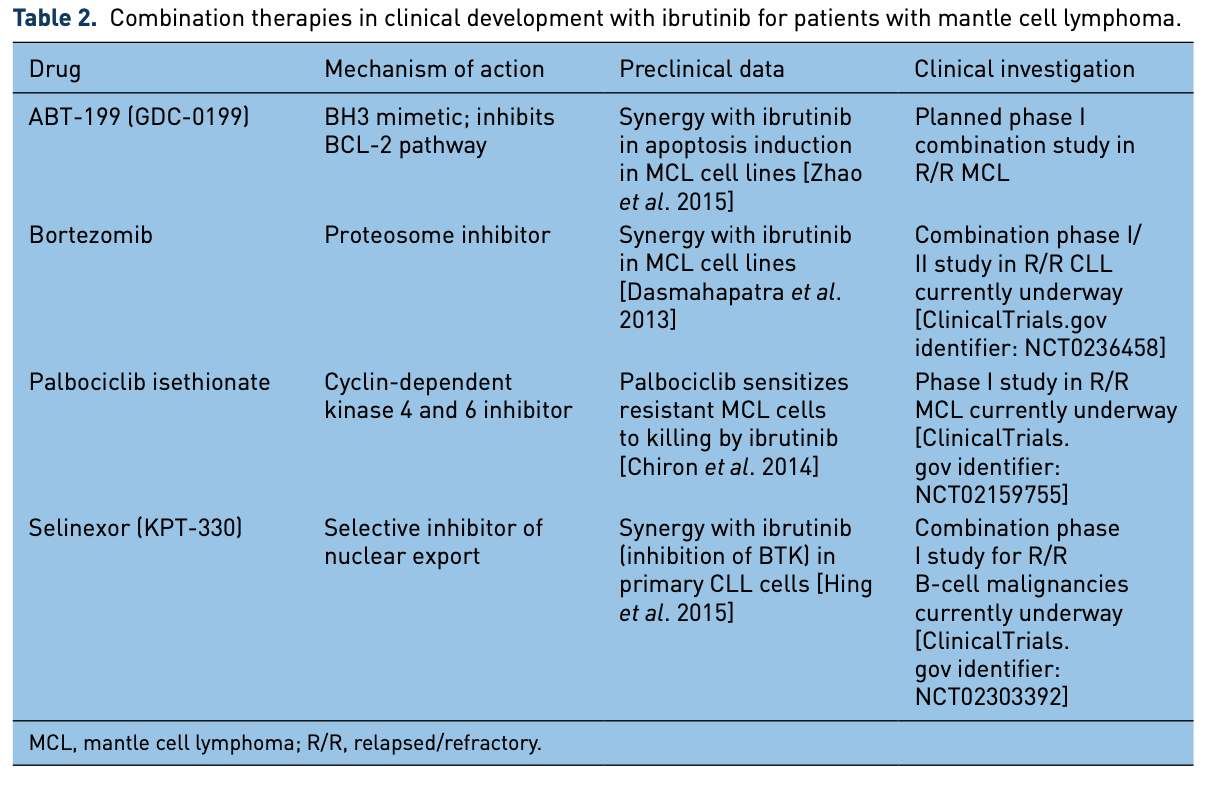
Multiple other combination clinical trials or trial concepts are in various stages of development and implementation, which are described in Table 2.
Pitfalls in the use of ibrutinib for patients
with MCL
Despite the notable clinical efficacy of ibrutinib in the treatment of patients with MCL, there are features about the drug that may affect the drug’s future development or use in the clinic, including cost, off-target effects, drug interactions, and development of resistance.
AEs related to off-target effects of ibrutinib
In addition to inhibition of BTK, ibrutinib can also inhibit other kinases with similar cysteine residues at their active sites, such as ITK, epidermal growth factor receptor (EGFR), tyrosine protein kinase (TEC), B-lymphocyte kinase (BLK), epithelial/ endothelial tyrosine kinase (BMX), human epider- mal growth factor 2 receptor (HER2), and human epidermal growth factor 4 (HER4), among others [Herrera and Jacobsen, 2014; Woyach et al. 2014], which may lead to off-target side effects. ITK nor- mally drives proximal T-cell receptor signaling and positively regulates NK cell’s Fc Receptor-initiated cytotoxicity via recognition of antibody-coated targets [Khurana et al. 2007]. Ibrutinib has been shown to irreversibly inhibit ITK [Dubovsky et al. 2013], which in preclinical models, has been shown to limit the therapeutic activity of monoclonal antibody therapies through ablation of antibody- dependent NK-cell-mediated cytotoxicity [Kohrt et al. 2014]. However, given the impressive clinical activity seen with ibrutinib in combination with rituximab [Wang et al. 2014a] this finding may not be clinically significant. Furthermore, this effect may not be as relevant in the novel Fc-engineered typeIIantibodiessuchasobinutuzumab[DaRoit et al. 2015]. In addition to potentially interfering
with the clinical efficacy of anti-CD20 therapy, inhibition of ITK may have additional clinical con- sequences. The majority of patients with a rare genetic ITK deficiency syndrome develop severe Epstein Barr virus infections that can lead to fatal lymphoproliferative disorders [Huck et al. 2009]. Althoughthiswarrantsmention,especiallyasmore patients are subjected to prolonged ITK inhibi- tion, to date, this has not been widely reported in patients receiving ibrutinib.
Ibrutinib has been shown to inhibit EGFR [Gao et al. 2014]. EGFR-inhibiting agents are currently used in the treatment of multiple types of solid tumors. As there are high levels of EGFR expres- sion in the basal layer of the epidermis [Nanney et al. 1984] and normal gastrointestinal mucosa [Hirsh et al. 2014], the most common AEs demon- strated by patients treated with EGFR-inhibiting agents are skin rashes and diarrhea. Thus, the skin rashes and diarrhea commonly seen in patients treated with ibrutinib are not surprising [Byrd et al. 2013, Wang et al. 2014b]. Similar to other EGFR inhibitors, ibrutinib-related rashes should be man- aged according to the severity of the reaction. For example, localized reactions with no physical symp- toms can be managed with topical low-potency steroid (1% hydrocortisone) or topical antibiotics (2% clindamycin). Moderate reactions (those with systemic symptoms) can be treated with topical treatmentsandtheadditionofanoraltetracycline antibiotic(doxycycline100mgdailyorminocycline 100mg twice daily for 4 weeks). Severe reactions should be managed with at least temporary discon- tinuation of the drug and use the preceding treat- ment options [Melosky and Hirsh, 2014]. Ibrutinib can potentially be restarted with a dose reduction if therashhasdecreasedtoatleastmoderateseverity. The other EGFR inhibitor-related side effect, diar- rhea, is most likely to occur in the first 4 weeks after treatment initiation, and importantly, the risk seems to diminish over time. Around half of ibrutinib- treated patients will experience diarrhea, the major- ity of which is grade 1–2.The diarrhea is thought to be primarily secretory, although the mechanism is unclear [Hirsh et al. 2014]. Similar to management of EGFR inhibitor related diarrhea, management of patients with ibrutinib-induced diarrhea should include patient education, nonpharmacologic man- agement strategies (including dietary change and increased fluid intake), pharmacologic manage- ment strategies (most commonly loperamide), or temporary drug discontinuation for grade 3 diar- rhea (at least seven stools daily or interfering with activities of daily living).
Other AEs reported in up to 50% of ibrutinib- treated patients were various severities of bleed- ing. Most events were grade 1/2 (spontaneous bruising or petechiae), but in 5% of the patients, the events were grade 3 or higher following trauma [Advani et al. 2013; Byrd et al. 2013; Wang et al. 2014b]. In vitro, ibrutinib has been shown to inhibit BTK and TEC kinase leading to inhibition of GPV1- and GP1b-mediated platelet function, which essentially inhibits platelet aggregation, resulting in decreased platelet adhesion on von Willebrand factor [Levade et al. 2014].The noted bleeding AEs in clinical trials have led to restric- tion of patients’ use of anticoagulants, specifically warfarin, while on ibrutinib studies. Importantly, since the majority of bleeding is minor, it can be managed without significant treatment interrup- tions or dose modifications. However, this side effect can be particularly concerning for patients, and as such, they should be counseled on this potential AE. Of note, although not reported in clinical studies, in our experience of using ibruti- nib, we have also seen bleeding in somewhat atypical sites, including recurrent spontaneous hemotypanum and hemothorax. Of note, patients with NHL and CLL are typically older and may require ongoing anticoagulation, which may limit the use of ibrutinib in these patients. No data exist detailing the safety of the combination of the new oral anticoagulants and ibrutinib. Therefore, whenever possible, anticoagulation should be avoided in patients on ibrutinib. However, if required, we preferentially use low-weight molec- ular heparin (typically enoxaparin) as it is short acting and its effects can be reversed in the event of bleeding. In patients without other viable clini- cal options, we have also successfully and safely treated a number of patients with oral direct thrombin inhibitors in combination with full-dose ibrutinib, noting that patients should be counse- led extensively on potential risks and this approach should be done with caution, especially in patients with underlying thrombocytopenia. In addition, although the use of antiplatelet agents, such as aspirin and clopidogrel, are not absolutely con- traindicated, we recommend discontinuing their use unless these agents are deemed absolutely necessary. In the event of a bleeding episode, platelet transfusion has been shown to correct aberrant ibrutinib-related hemostasis [Levade et al. 2014]. If a patient requires minor or major surgery, we follow the package insert which rec- ommends abstention from ibrutinib for 3 to 7 days before and after a minor or major procedure, respectively.
Another AE that has been attributed to ibrutinib in multiple clinical trials which was most notable in the RESONATE study, a phase III clinical trial comparing ibrutinib with ofatumumab (anti- CD20 monoclonal antibody) in patients with CLL [Byrd et al. 2014] is afib. Five percent of patients receiving ibrutinib (3% were grade ⩾ 3) developed afib compared with less than 1% of patients who received ofatumumab (none were grade ⩾ 3).The mechanisms for this AE have not been well defined; however, a preclinical mouse model suggested that afib may be induced by inhibition of phosphatidylinositol 3-kinase p110-α (PI3K; another important member of the BCR signaling pathway) in cardiac myocytes [McMullen et al. 2014].The authors of this study concluded that the patients who received ibruti- nib on RESONATE may have been at a higher baseline risk for afib. Accordingly, although a fur- ther understanding of the potential risks of afib with ibrutinib is needed, afib is not an absolute contraindication to treatment with ibrutinib, and the development of afib does not require discon- tinuation of ibrutinib as the risk usually does not outweigh ibrutinib’s clinical benefit. However, given potential drug interactions, pharmacologic cardioversion in patients receiving ibrutinib should only be considered after discussions with a pharmacist and such patients should be moni- tored closely. As mentioned previously, if a patient requires anticoagulation for afib, one should pro- ceed with caution.
Studies using ibrutinib have not extensively evalu- ated interruption of therapy. Instead, studies have required continuous dosing even after remission has been achieved. Given the potential toxicities described above as well as the financial burden (>$10,000 per month) of ongoing therapy, studies are needed to evaluate shorter durations of ther- apy, depth of response (i.e. ability to achieve mini- mal residual disease negativity), and planned dose interruptions in responding patients. Secondary to the clinical impact of off-target side effects of ibrutinib, second generation and more specific BTK inhibitors, such as ACP-196, have been developedandareinvariousstagesofclinicaltrials. [ClinicalTrials.gov identifier: NCT02213926], CC-292, and ONO-4059 [ClincalTrials.gov iden- tifier: NCT01659255]. Although at an earlier stage of clinical development, CC-292 treatment has resulted in similar toxicity profile with diar- rhea as the most common AE [Brown et al. 2013]. Results for clinical trials with ACP-196 have yet to be reported to demonstrate if improved binding
specificity will ablate some of the off-target AEs seen with ibrutinib without compromising efficacy.
Drug interactions with ibrutinib
Drug interactions must also be analyzed carefully when considering ibrutinib treatment. The drug is metabolized in the liver by cytochrome P450 3A4 (CYP3A4), which can result in drug interac- tions with inducers or inhibitors of CYP3A4 [Herrera and Jacobsen, 2014]. Notable inhibitors of CYP3A4 are grapefruit juice, azole antifungal agents, anti-human immunodeficiency virus agents, metronidazole, ciprofloxacin, and tetracy- cline. Notable inducers of CYP3A4 are antisei- zure medications (carbamazepine, oxycarbazepine, fosphenytoin, phenytoin, pentobarbital, and phe- nobarbital). Prior to initiation of ibrutinib, a care- ful review of current medications with changes to potential interacting drugs is required for patient safety.
Relapses after and refractoriness to ibrutinib
Acquired and primary resistance have been seen in patients with MCL treated with ibrutinib, and these patients have demonstrated poor clinical outcomes. Similar to chronic myelogenous leuke- mia, where acquired resistance can be seen after prolonged tyrosine kinase inhibitor therapy [O’Dwyer et al. 2004], prolonged therapy with ibrutinib can lead to resistance via prevention of irreversible binding of BTK. For example, Chiron and colleagues evaluated serial biopsies in ibruti- nib-treated patients with MCL. Two of seven patients with acquired resistance to ibrutinib (who had attained PR on therapy with duration of response of 14 and 30 months, respectively), were found to have a cysteine to serine mutation at the BTK binding site of ibrutinib (BTKC481S), which decreases ibrutinib’s binding affinity, rendering it ineffective [Chiron et al. 2014]. Importantly, these mutations were not identified prior to ibru- tinib treatment. The same mutation was first described in patients with CLL with acquired resistance to ibrutinib [Woyach etal. 2014]. Conversely, the BTKC481S mutation was not found in six patients with MCL with primary or early acquired resistance to ibrutinib (on therapy for < 5 months) [Chiron et al. 2014]. In these patient samples, the investigators found that downstream kinases in the BCR signaling path- way were activated, specifically PI3K and protein kinase B (AKT). Similarly, another group found
that in MCL cell lines, ibrutinib resistance may be caused by activation of the alternative nuclear factor light-chain enhancer of activated B cells (NFκB) pathway (via MAP3K14), whereas the pathogenesis of ibrutinib-sensitive MCL cell lines depends upon the classical BCR-BTK-NFκB pathway [Rahal et al. 2014]. Patient data support the role of the NFκB pathway in primary ibruti- nib resistance. In a phase II study in which 110 patients with MCL R/R after rituximab contain- ing chemotherapy and bortezomib therapy received single-agent ibrutinib (SPARK Trial) [ClinicalTrials.gov identifier: NCT01599949], 25 patients were considered to have primary treatment resistance and another 22 patients were classified as having moderate clinical benefit defined as SD (n=16) or response lasting less than 12 months (n = 6). Interestingly, only 20% of the patients with primary resistance were deemed to have blastic MCL, an aggressive subtype of MCL associated with poor outcomes, while 38% had a high mantle cell international prognostic index. In an attempt to identify potential genetic aberrations associated with ibrutinib resistance, DNA was extracted from pretreatment tumor samples and enriched libraries were constructed with probe sets specific for the coding region of 97 genes possibly involved in ibrutinib response and resistance. Deep sequencing was performed and aligned to the reference genome. A total of 27 genes were found with nonsynonymous variants in at least two patients. No mutations previously described in patients with MCL or CLL and acquired resistance to ibrutinib were seen. Genes previously implicated in diffuse large B-cell lymphoma (DLBCL) pathogenesis such as mixed lineage leukemia protein 2 (MLL2) and cAMP response element binding protein (CREBBP) were mutated along with mutation in proto- oncogene serine/threonine-protein kinase (PIM1) and erb-b2 receptor tyrosine kinase 4 (ERBB4) kinase genes. Several of the mutations detected affect NFκB signaling inhibition, which is an inte- gral part of the BCR signaling pathway, normally inhibited by ibrutinib [Balasubramanian etal. 2014]. These results are hypothesis generating and may implicate important signaling pathways; how- ever, whether the observed mutations are markers of poor risk or drivers of resistance remains unclear.
The outcome of patients with MCL with acquired resistance to ibrutinib is poor. One study describes 42 patients with MCL who discontinued therapy with ibrutinib. Among 31 patients who experi- enced disease progression following ibrutinib and
underwent salvage therapy, the ORR was only 32%. At a median follow up of 10.7 months (range 2.4– 38.9 months), the median OS after disease progres- sion was only 8.4 months [Cheah et al. 2015]. Another retrospective, multi-institutional cohort studydescribed32patientswithMCLwhoexperi- enced disease progression while receiving ibrutinib. Of the 19 patients who received a salvage therapy after ibrutinib, 17 were evaluable for response and only 6 of those patients (35%) achieved an objec- tiveresponsetosubsequenttreatment.Themedian OS following cessation of ibrutinib was only 4 months (95% CI 2–10 months) [Martin et al. 2014b]. Analysis of a larger international multi- center cohort of MCL ibrutinib failures is ongoing. Thesedataareinlinewithwhatwehaveseeninthe clinic, including the development of aggressive dis- ease and blastoid transformation while on therapy. It is not yet clear if the poor outcomes seen after ibrutinib failure simply represent the final common pathway of treating patients with advanced MCL or the result of ibrutinib selecting for a biologically more aggressive phenotype, the latter of which could have significant implications given the imple- mentation of ibrutinib in the frontline treatment setting. Further studies to better understand ibruti- nib, the mechanisms driving resistance, and how to treat these patients are urgently needed.
Summary
This article details the clinical experience with ibrutinib in the treatment of patients with MCL, including completed and published clinical trials and a review of potential AEs and resistance. Ibrutinib is a promising agent for patients with MCL with notable response rates. However, as a singleagent,nearlyonethirdofpatientsrelapsein the first 2 years of treatment, which calls for inves- tigation of combination therapies as well as evalu- ating the addition of ibrutinib to upfront treatment strategies. Although most AEs experienced by patients treated with ibrutinib are mild, some can be severe and treatment limiting and may be attributed to off-target effects of the drug. Therefore, attempts to develop more specific sec- ond-generation BTK inhibitors that may mitigate some of these AEs are underway. Prior to prescrib- ing ibrutinib, a thorough review of a patient’s medications should be completed to determine potential drug interactions. Caution should be used when considering ibrutinib for patients requiring chronic anticoagulation, noting that additional studies are needed to better character- ize risks in patients on newer anticoagulants.
Patients who develop resistance to ibrutinib dem- onstrate very poor clinical outcomes. Increasingly, as ibrutinib is studied earlier in the course of the disease, special attention to progression patterns, disease biology, and outcomes after ibrutinib fail- ure will be paramount. Further definition of resist- ance and investigation of strategies to improve clinical outcomes in these patients remain an area of high unmet clinical need. Evaluation of second- generation BTK inhibitors is ongoing. Studies of the impact of shorter treatment duration, effects on minimal residual disease, initiation of therapy on molecular relapse, and incorporation of novel combinations are warranted.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not- for-profit sectors.
Conflict of interest statement
Dr. Spurgeon: Research funding: Pharmacyclics, Janssen Pharmaceuticals Inc., and Acerta Pharma. Honoraria: Pharmacyclics.
References
Advani, R., Buggy, J., Sharman, J., Smith, S., Boyd, T., Grant, B. et al. (2013) Bruton tyrosine kinase inhibitor ibrutinib (PCI-32765) has significant activity in patients with relapsed/refractory B-cell malignancies. J Clin Oncol 31: 88–94.
Balasubramanian, S., Schaffer, M., Deraedt, W., Davis, C., Stepanchick, E., Aquino, R. et al. (2014) Mutational analysis of patients with primary resistance to single-agent ibrutinib in relapsed or refractory mantle cell lymphoma (MCL). Blood (ASH Annual Meeting Abstracts) abstract 78.
Bojarczuk, K., Siernicka, M., Dwojak, M., Bobrowicz, M., Pyrzynska, B., Gaj, P. et al. (2014) B-cell
receptor pathway inhibitors affect CD20 levels and impair antitumor activity of anti-CD20 monoclonal antibodies. Leukemia 28: 1163–1167.
Brown, J., Harb, W., Hill, B., Gabrilove, J., Sharman, J., Schreeder, M. et al. (2013) Phase 1 study of single agent CC-292, a highly selective Bruton’s tyrosine kinase (BTK) inhibitor, in relapsed/refractory chronic lymphocytic leukemia (CLL). Blood (ASH Annual Meeting Abstracts) 122:abstract 3793.
Byrd, J., Brown, J., O’Brien, S., Barrientos, J., Kay, N., Reddy, N. et al. (2014) Ibrutinib versus of atumumab in previously treated chronic lymphoid leukemia. N Engl J Med 371: 213–223.
Byrd, J., Furman, R., Coutre, S., Flinn, I., Burger, J., Blum, K. et al. (2013) Targeting BTK with ibrutinib
in relapsed chronic lymphocytic leukemia. N Engl J Med 369: 32–42.
Chandran, R., Gardiner, S., Simon, M. and Spurgeon, S. (2012) Survival trends in mantle cell lymphoma in the United States over 16 years 1992- 2007. Leuk Lymphoma 53: 1488–1493.
Cheah, C., Chihara, D., Romaguera, J., Fowler, N., Seymour, J., Hagemeister, F. et al. (2015) Patients with mantle cell lymphoma failing ibrutinib are unlikely to respond to salvage chemotherapy and have poor outcomes. Ann Oncol 26: 1175–1179.
Chiron, D., Di Liberto, M., Martin, P., Huang, X., Sharman, J., Blecua, P. et al. (2014) Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed
by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov 4: 1022–1035.
Christian, B., Kuruvilla, J., Smith, S., Porcu, P., Ruppert, A., Byrd, J. et al. (2014) A phase I study of ibrutinib and lenalidomide in patients with relapsed and refractory B-cell non-Hodgkin’s lymphoma. Blood (ASH Annual Meeting Abstracts) abstract 4476.
Cinar, M., Hamedani, F., Mo, Z., Cinar, B., Amin, H. and Alkan, S. (2013) Bruton tyrosine kinase is commonly over expressed in mantle cell lymphoma and its attenuation by Ibrutinib induces apoptosis. Leuk Res 37: 1271–1277.
Da Roit, F., Engelberts, P., Taylor, R., Breij, E., Gritti, G., Rambaldi, A. et al. (2015) Ibrutinib interferes with the cell-mediated anti-tumour activities of therapeutic CD20 antibodies: implications for combination therapy. Haematologica 100: 77–86.
Dasmahapatra, G., Patel, H., Dent, P., Fisher, R., Friedberg, J. and Grant, S. (2013) The Bruton tyrosine kinase (BTK) inhibitor PCI-32765 synergistically increases proteasome inhibitor activity in diffuse large-B cell lymphoma (DLBCL) and mantle cell lymphoma (MCL) cells sensitive or resistant to bortezomib. Br J Haematol 161: 43–56.
Dubovsky, J., Beckwith, K., Natarajan, G., Woyach, J., Jaglowski, S., Zhong, Y. et al. (2013) Ibrutinib is an irreversible molecular inhibitor of ITK driving a Th1-selective pressure in T lymphocytes. Blood 122: 2539–2549.
Flinn, I., van der Jagt, R., Kahl, B., Wood,
P., Hawkins, T., Macdonald, D. et al. (2014) Randomized trial of bendamustine-rituximab or R-CHOP/R-CVP in first-line treatment of indolent NHL or MCL: the BRIGHT study. Blood 123: 2944–2952.
Gao, W., Wang, M., Wang, L., Lu, H., Wu, S.,
Dai, B. et al. (2014) Selective antitumor activity of ibrutinib in EGFR-mutant non-small cell lung cancer cells. J Natl Cancer Inst 106. doi:10.1093/jnci/dju204. Print 2014 Sep.
Geisler, C., Kolstad, A., Laurell, A., Andersen, N., Pedersen, L., Jerkeman, M. et al. (2008) Long-term progression-free survival of mantle cell lymphoma after intensive front-line immunochemotherapy with in vivo-purged stem-cell rescue: a nonrandomized phase 2 multicenter study by the Nordic Lymphoma Group. Blood 112: 2687–2693.
Geisler, C., Kolstad, A., Laurell, A., Jerkeman,
M., Raty, R., Andersen, N. et al. (2012) Nordic MCL2 trial update: six-year follow-up after intensive immunochemotherapy for untreated mantle cell lymphoma followed by BEAM or BEAC + autologous stem-cell support: still very long survival but late relapses do occur. Br J Haematol 158: 355–362.
Goy, A., Sinha, R., Williams, M., Kalayoglu Besisik, S., Drach, J., Ramchandren, R. et al. (2013) Single- agent lenalidomide in patients with mantle-cell lymphoma who relapsed or progressed after or
were refractory to bortezomib: phase II MCL-001 (EMERGE) study. J Clin Oncol 31: 3688–3695.
Habermann, T., Lossos, I., Justice, G., Vose, J., Wiernik, P., McBride, K. et al. (2009) Lenalidomide oral monotherapy produces a high response rate
in patients with relapsed or refractory mantle cell lymphoma. Br J Haematol 145: 344–349.
Herrera, A. and Jacobsen, E. (2014) Ibrutinib for the treatment of mantle cell lymphoma. Clin Cancer Res 20: 5365–5371.
Herrmann, A., Hoster, E., Zwingers, T., Brittinger, G., Engelhard, M., Meusers, P. et al. (2009) Improvement of overall survival in advanced stage mantle cell lymphoma. J Clin Oncol 27: 511–518.
Hing, Z., Mantel, R., Beckwith, K., Guinn, D., Williams, E., Smith, L. et al. (2015) Selinexor is effective in acquired resistance to ibrutinib and synergizes with ibrutinib in chronic lymphocytic leukemia. Blood 125: 3128–3132.
Hirsh, V., Blais, N., Burkes, R., Verma, S. and Croitoru, K. (2014) Management of diarrhea induced by epidermal growth factor receptor tyrosine kinase inhibitors. Curr Oncol 21: 329–336.
Honigberg, L., Smith, A., Sirisawad, M., Verner,
E., Loury, D., Chang, B. et al. (2010) The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc Natl Acad Sci USA 107: 13075–13080.
Huck, K., Feyen, O., Niehues, T., Ruschendorf, F., Hubner, N., Laws, H. et al. (2009) Girls homozygous for an IL-2-inducible T cell kinase mutation that leads to protein deficiency develop fatal EBV-associated lymphoproliferation. J Clin Invest 119: 1350–1358.
Khurana, D., Arneson, L., Schoon, R., Dick, C. and Leibson, P. (2007) Differential regulation of human
NK cell-mediated cytotoxicity by the tyrosine kinase ITK. J Immunol 178: 3575–3582.
Kluin-Nelemans, H., Hoster, E., Hermine, O., Walewski, J., Trneny, M., Geisler, C. et al. (2012) Treatment of older patients with mantle-cell lymphoma. N Engl J Med 367: 520–531.
Kohrt, H., Sagiv-Barfi, I., Rafiq, S., Herman, S., Butchar, J., Cheney, C. et al. (2014) Ibrutinib antagonizes rituximab-dependent NK cell-mediated cytotoxicity. Blood 123: 1957–1960.
Le Gouill, S., Thieblemont, C., Oberic, L., Bouabdallah, K., Gyan, E., Damaj, G. et al. (2014) Rituximab maintenance versus wait and watch after four courses of R-DHAP followed by autologous stem cell transplantation in previously untreated young patients with mantle cell lymphoma: first interim analysis of the phase III prospective lyma trial. Blood (ASH Annual Meeting Abstracts) abstract 146.
Levade, M., David, E., Garcia, C., Laurent, P., Cadot, S., Michallet, A. et al. (2014) Ibrutinib treatment affects collagen and von Willebrand factor- dependent platelet functions. Blood 124: 3991–3995.
Maddocks, K., Christian, B., Jaglowski, S., Flynn, J., Jones, J., Porcu, P. et al. (2015) A phase 1/1b study of rituximab, bendamustine, and ibrutinib in patients with untreated and relapsed/refractory non-Hodgkin lymphoma. Blood 125: 242–248.
Martin, P., Goy, A., Ramchandren, R., Ferrante, L., Reddy, V., Londhe, A. et al. (2014a) Safety Results from the United States Cohort of the ibrutinib
early access treatment protocol (EAP: MCL4001)
in patients with relapsed or refractory mantle cell lymphoma. Blood (ASH Annual Meeting Abstracts) abstract 4461.
Martin, P., Maddocks, K., Noto, K., Christian, B., Furman, R., Andritsos, L. et al. (2014b) Poor overall survival of patients with ibrutinib-resistant mantle cell lymphoma. Blood (ASH Annual Meeting Abstracts) abstract 3047.
McMullen, J., Boey, E., Ooi, J., Seymour, J., Keating, M. and Tam, C. (2014) Ibrutinib increases the risk
of atrial fibrillation, potentially through inhibition of cardiac PI3K-Akt signaling. Blood 124: 3829–3830.
Melosky, B. and Hirsh, V. (2014) Management of common toxicities in metastatic NSCLC related to anti-lung cancer therapies with EGFR-TKIs. Front Oncol 4: 238.
Merli, F., Luminari, S., Ilariucci, F., Petrini, M., Visco, C., Ambrosetti, A. et al. (2012) Rituximab plus HyperCVAD alternating with high dose cytarabine and methotrexate for the initial treatment of patients with mantle cell lymphoma, a multicentre trial from Gruppo Italiano Studio Linfomi. Br J Haematol 156: 346–353.
Nanney, L., Magid, M., Stoscheck, C. and King, L., Jr (1984) Comparison of epidermal growth factor binding and receptor distribution in normal human epidermis and epidermal appendages. J Invest Dermatol 83: 385–393.
O’Dwyer, M., Mauro, M., Blasdel, C., Farnsworth, M., Kurilik, G., Hsieh, Y. et al. (2004) Clonal evolution and lack of cytogenetic response are adverse prognostic factors for hematologic relapse of chronic phase CML patients treated with imatinib mesylate. Blood 103: 451–455.
Pighi, C., Gu, T., Dalai, I., Barbi, S., Parolini, C., Bertolaso, A. et al. (2011) Phospho-proteomic analysis of mantle cell lymphoma cells suggests a pro-survival role of B-cell receptor signaling. Cell Oncol (Dordr) 34: 141–153.
Rahal, R., Frick, M., Romero, R., Korn, J., Kridel, R., Chan, F. et al. (2014) Pharmacological and genomic profiling identifies NF-kappaB-targeted treatment strategies for mantle cell lymphoma. Nat Med 20: 87–92.
Rinaldi, A., Kwee, I., Taborelli, M., Largo, C., Uccella, S., Martin, V. et al. (2006) Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol 132: 303–316.
Robak, T., Huang, H., Jin, J., Zhu, J., Liu, T., Samoilova, O. et al. (2015) Bortezomib-based therapy for newly diagnosed mantle-cell lymphoma. N Engl J Med 372: 944–953.
Romaguera, J., Fayad, L., Feng, L., Hartig, K., Weaver, P., Rodriguez, M. et al. (2010) Ten-year follow-up after intense chemoimmunotherapy with Rituximab-HyperCVAD alternating with Rituximab- high dose methotrexate/cytarabine (R-MA) and without stem cell transplantation in patients with untreated aggressive mantle cell lymphoma. Br J Haematol 150: 200–208.
Rummel, M., Niederle, N., Maschmeyer, G., Banat, G., von Grunhagen, U., Losem, C. et al. (2013) Bendamustine plus rituximab versus CHOP plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: an open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet 381: 1203–1210.
Sharman, J., Farber, C., Mahadevan, D., Schreeder, M., Brooks, H., Kolibaba, K. et al. (2014) Ublituximab (TG-1101), a novel glycoengineered anti-CD20 monoclonal antibody, in combination with ibrutinib is highly active in patients with relapsed and/
or refractory CLL and MCL; results of a phase II trial. Blood (ASH Annual Meeting Abstracts) abstract 4679.
Wang, M., Hagemeister, F., Westin, J., Fayad, L., Samaniego, F., Turturro, F. et al. (2014a) Ibrutinib and rituximab are an efficacious and safe combination in relapsed mantle cell lymphoma: preliminary results from a phase II clinical trial. Blood (ASH Annual Meeting Abstracts) abstract 627.
Wang, M., Rule, S., Martin, P., Goy, A., Auer, R., Kahl, B. et al. (2013) Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369: 507–516.
Wang, M., Rule, S., Martin, P., Goy, A., Auer, R., Kahl, B. et al. (2014b) Single-agent ibrutinib demonstrates safety and durability of response
at 2 years follow-up in patients with relapsed or refractory mantle cell lymphoma: updated results of an international, multicenter, open-label phase 2 study. Blood (ASH Annual Meeting Abstracts) abstract 4453.
Woyach, J., Furman, R., Liu, T., Ozer, H., Zapatka, M., Ruppert, A. et al. (2014) Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 370: 2286–2294.
Younes, A., Thieblemont, C., Morschhauser,
F., Flinn, I., Friedberg, J., Amorim, S. et al.
(2014) Combination of ibrutinib with rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) for treatment-naive patients with CD20-positive B-cell non-Hodgkin lymphoma: a non-randomised, phase 1b study. Lancet Oncol 15: 1019–1026.
Zhao, X., Bodo, J., Sun, D., Durkin, L., Lin, J., Smith, M. et al. (2015) Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. Br J Haematol 168: 765–768.
Zhou, Y., Wang, H., Fang, W., Romaguer, J., Zhang, Y., Delasalle, K. et al. (2008) Incidence trends of mantle cell lymphoma in the United States between 1992 and 2004. Cancer 113: 791–798.
Zinzani, P., Vose, J., Czuczman, M., Reeder, C., Haioun, C., Polikoff, J. et al. (2013) Long-term follow-up of lenalidomide in relapsed/refractory mantle cell lymphoma: subset analysis of the NHL- 003 study. Ann Oncol 24: 2892–2897.
